(一)發病原因
引起肝硬化的病因很多,不同地區的主要病因也不相同。歐美以酒精性肝硬化為主,我國以肝炎病毒性肝硬化多見,其次為血吸蟲病肝纖維化,酒精性肝硬化亦逐年增加。研究證實,2種病因先後或同時作用於肝髒,更易產生肝硬化。如血吸蟲病或長期大量飲酒者合並乙型病毒性肝炎等。
1.肝炎後肝硬化(posthepatitic cirrhosis) 指病毒性肝炎發展至後期形成肝硬化。現已知肝炎病毒有甲、乙、丙、丁、戊等類型。近年研究認為甲型肝炎及戊型肝炎無慢性者,除急性重症外,不形成肝硬化。乙、丙型肝炎容易轉成慢性即慢性活動性肝炎和肝硬化。
1974年Shikatu報道用免疫熒光方法可以顯示HBsAg(乙型肝炎表面抗原)。在顯微鏡下含有HBsAg的細胞漿呈毛玻璃狀,用地衣紅(Orecein)染色法可將含HBsAg的肝細胞漿染成光亮的橘紅色。經過多年保存的肝硬化標本,用此法也可顯示出來含HBsAg的肝細胞,使乙型肝炎病毒引起的肝硬化有了可靠的依據。乙肝病人10%~20%呈慢性經過,長期HBsAg陽性,肝功能間歇或持續異常。乙肝病毒在肝內持續復制可使淋巴細胞在肝內浸潤,釋放大量細胞因子及炎性介質,在清除病毒的同時使肝細胞變性、壞死,病變如反復持續發展,可在肝小葉內形成纖維隔、再生結節而形成肝硬化。68%的丙型肝炎呈慢性過程,30%的慢性丙型肝炎發展為肝硬化。丁型肝炎可以和乙型肝炎同時感染或重疊感染,可以減慢乙型肝炎病毒的復制,但常加劇病變的活動及加速肝硬化的發生。
病毒性肝炎的急性重症型,肝細胞大塊壞死融合,從小葉中心向匯管區擴展,引起網狀支架塌陷、靠攏、形成纖維隔。並產生小葉中心至匯管區的架橋現象,而形成大結節性肝硬化。慢性活動性肝炎形成的肝硬化,在匯管區有明顯的炎症和纖維化,形成寬的不規則的“主動性”纖維隔,向小葉內和小葉間伸展,使鄰接的各小葉被纖維隔分隔、破壞。這時雖然肝髒結構被改建,但還不是肝硬化,而是肝纖維化階段。當炎症從肝小葉邊緣向中心部擴展,引起點片狀壞死和單核細胞浸潤,纖維隔也隨之繼續向中心擴展,分割肝小葉,加之肝細胞再生,形成被結締組織包繞的再生結節,則成為肝硬化。至病變的末期,炎症和肝細胞壞死可以完全消失,只是在纖維隔中有多數大小不等的結節,結節為多小葉性,形成大結節性肝硬化。如肝炎病變較輕,病程進行較慢,也可以形成小結節性肝硬化、混合性肝硬化或再生結節不明顯性肝硬化(不完全分隔性肝硬化)。
從病毒性肝炎發展為肝硬化,據研究表明與感染抗原量無關。而與病毒毒力及人體免疫狀態有顯著關系。遺傳因素與慢性化傾向有關,與人類白細胞抗原HL-A1、HL-A8缺乏似有關系,但尚待進一步研究。
2.酒精性肝硬化(alcoholic cirrhosis) 西方國家酒精性肝硬化發病率高,由酗酒引起。近年我國酒的消耗量增加,脂肪肝及酒精性肝硬化的發生率也有所增高。據統計肝硬化的發生與飲酒量和時間長短成正比。每天飲含酒精80g的酒即可引起血清谷丙轉氨酶升高,持續大量飲酒數周至數月多數可發生脂肪肝或酒精性肝炎。若持續大量飲酒達15年以上,75%可發生肝硬化。
酒精進入肝細胞後,在乙醇脫氫酶和微粒體乙醇氧化酶作用下,轉變為乙醛,乙醛再轉變為乙酸,乙酸使輔酶Ⅰ(NAD)過多的轉變為還原型輔酶Ⅰ(NADH)因而NAD減少,NADH增加,則兩者比值下降,線粒體內三羧酸循環受到抑制,脂肪酸的酯化增加,三酰甘油增加,肝內的三酰甘油釋放減少,另外肝內NADH過多,又促進了脂肪酸的合成,使體脂肪形成脂肪酸的動力加強造成肝內三酰甘油過多,超過肝髒的處理能力,而發生脂肪肝。長期大量飲酒,可使肝細胞進一步發生變性、壞死和繼發炎症,在脂肪肝的基礎上發生酒精性肝炎,顯微鏡下可見肝細胞廣泛變性和含有酒精性透明蛋白(Mallorys alcoholic hyalin)匯管區有多形核白細胞及單核細胞浸潤和膽小管增生,纖維組織增生,最後形成小結節性肝硬化。酒精性肝硬化小葉中心靜脈可以發生急性硬化性透明樣壞死引起纖維化和管腔閉塞,加劇門靜脈高壓。中心部纖維化向周緣部位擴展,也可與匯管區形成“架橋”現象。
3.寄生蟲性肝硬化(parasitic cirrhosis) 如血吸蟲或肝吸蟲等蟲體在門脈系統寄居,蟲卵隨門脈血流沉積於肝內,引起門靜脈小分支栓塞。蟲卵大於肝小葉門靜脈輸入分支的直徑,故栓塞在匯管區引起炎症、肉芽腫和纖維組織增生,使匯管區擴大,破壞肝小葉界板,累及小葉邊緣的肝細胞。肝細胞再生結節不明顯,可能與蟲卵堵塞門靜脈小分支,肝細胞營養不足有關。因門靜脈受阻,門脈高壓症明顯,有顯著的食管靜脈曲張和脾大。成蟲引起細胞免疫反應和分泌毒素,是肝內肉芽腫形成的原因。蟲卵引起體液免疫反應,產生抗原-抗體復合物,可能是肝內門脈分支及其周圍發生炎症和纖維化的原因。寄生蟲性肝硬化在形態學上屬再生結節不顯著性肝硬化。
4.中毒性肝硬化(toxic cirrhosis) 化學物質對肝髒的損害可分兩類:一類是對肝髒的直接毒物,如四氯化碳、甲氨蝶呤等;另一類是肝髒的間接毒物,此類毒物與藥量無關,對特異素質的病人先引起過敏反應,然後引起肝髒損害。少數病人可引起肝硬化,如異煙酰、異丙肼(iproniazid)、氟烷。其病變與肝炎後肝硬化相似。四氯化碳為肝髒的直接毒物,對肝髒的損害與藥量的大小成正比關系,引起肝髒彌漫性的脂肪浸潤和小葉中心壞死。四氯化碳本身不是毒性物質,經過藥物代謝酶的作用,如P-450微粒體酶系統,將四氯化碳去掉一個氯原子,而形成三氯甲烷,即氯仿,則成為肝細胞內質網和微粒體的藥物代謝酶系統的劇毒(產生三氯甲基自由基和氯自由基),引起肝細胞生物膜的脂質過氧化及肝細胞損害。由於對肝細胞內微細結構的破壞、藥物代謝酶減少又降低了對四氯化碳的代謝,從而減弱了對肝髒的繼續損害。病人在恢復之後,肝功能多能恢復正常。僅在反復或長期暴露在四氯化碳中才偶有發生大結節性肝硬化。
動物試驗反復給大鼠四氯化碳,使藥物蓄積可引起肝硬化。
氨甲喋呤是抗葉酸藥物,臨床常用以治療白血病、淋巴瘤、牛皮癬(銀屑病)等。據報道可引起小結節性肝硬化。
5.膽汁性肝硬化(biliary cirrhosis) 原發性膽汁性肝硬化(primary biliary cirrhosis)的原因和發病機制尚不清楚,可能與自身免疫有關。繼發性膽汁性肝硬化是各種原因的膽管梗阻引起,包括結石、腫瘤、良性狹窄及各種原因的外壓和先天、後天的膽管閉塞。多為良性疾病引起。因為惡性腫瘤多在病人發生肝硬化之前死亡。
各種原因引起的完全性膽管梗阻,病程在3~12個月方能形成肝硬化。發生率約占這類病人的10%以下。
膽管梗阻的早期,膽汁顏色變暗,但很快可變為白色。因膽汁淤積和膽管擴張,膽管內壓力增高,抑制膽汁分泌,膽汁可以由綠色變為白色,形成所謂“白膽汁”。顯微鏡下可見匯管區小膽管高度擴張,甚至膽管破裂,膽汁溢出使匯管區和肝小葉周緣區發生壞死及炎症,壞死灶被膽管溢出的膽汁所充滿,形成“膽池”。這是機械性膽管梗阻的一個特征表現。病變繼續進展,周緣區的壞死和炎症刺激使匯管區的纖維組織增生,並向小葉間伸延形成纖維隔。各匯管區的纖維隔互相連接,將肝小葉分割,呈不完全分隔性肝硬化。與肝炎後肝硬化、酒精性肝硬化的中心至匯管區纖維隔不同。但病變如繼續發展,到晚期也可出現匯管區至小葉中心區的纖維隔及肝細胞再生結節,而失去其特征性的表現,以致在病理形態上和臨床表現上與其他肝硬化不易區別。也可以出現門靜脈高壓及腹水。
膽管梗阻形成肝硬化的原理可能是由於肝內血管受到擴大膽管的壓迫及膽汁外滲,肝細胞發生缺血壞死。纖維組織向膽管伸展包圍小葉,並散布於肝細胞間,最後形成肝硬化。不完全性膽管梗阻很少發展為膽汁性肝硬化。
已知膽管感染不是形成肝硬化的必需條件。據報道,無感染的完全性膽管梗阻發展為膽汁性肝硬化者更為多見。
6.循環障礙(淤血)性肝硬化 由於各種心髒病引起的慢性充血性心力衰竭、縮窄性心包炎等使肝髒長期處於淤血和缺氧狀態,最終形成肝硬化。Budd-chiari綜合征是由於肝靜脈慢性梗阻造成長期肝淤血,也發生與心源性完全相同的肝硬化。
心功能不全時,由於心髒搏血量減少,肝內血液灌注下降,肝小葉邊緣部位血含氧量較高,流向肝小葉中心時,氧含量進行性減低。心功能不全同時又存在中心靜脈壓增高,中心靜脈及其周圍肝窦擴張、淤血、壓迫肝細胞,肝細胞變性、萎縮、甚至出血壞死。缺氧及壞死均可刺激膠原增生、發生纖維化,甚至發生中心靜脈硬化纖維化,逐漸由中心向周圍擴展,相鄰小葉的纖維素彼此聯結,即中心至中心的纖維隔。而匯管區相對受侵犯較少。這是循環障礙性肝硬化的特點。後期由於門脈纖維化繼續進展,肝實質壞死後不斷再生以及膽管再生則最後失去淤血性肝硬化特點。此型肝硬化在病理形態上呈小結節性或不完全分隔性肝硬化。
7.營養不良性肝硬化(malnutritional cirrhosis) 長期以來認為營養不良可以引起肝硬化。但一直缺乏直接證據。動物實驗予缺少蛋白質、膽鹼和維生素的飲食可以造成肝硬化的改變,但病變是可逆的,且缺少肝硬化病人常有的血管方面的繼發性變化。有的作者觀察了惡性營養不良(Kwashiorkor)病人,發現他們的肝損害是脂肪肝,並不發生肝硬化。僅兒童偶爾肝髒有彌漫性纖維增生,像似肝硬化,當給以富有蛋白質的飲食後,病變可以逆轉而肝髒恢復正常,只在某些病例可有輕度纖維增生。所以至今營養不良能否直接引起肝硬化還不能肯定。多數認為營養失調降低了肝髒對其他致病因素的抵抗力,如慢性特異性或非特異性腸炎除引起消化、吸收和營養不良外,病原體在腸內產生的毒素經門靜脈入肝,肝髒不能將其清除,而導致肝細胞變性壞死形成肝硬化。故此認為營養不良是產生肝硬化的間接原因。又如小腸旁路手術後引起的肝硬化,有人認為是由於營養不良,缺乏基本的氨基酸或維生素E,飲食中糖類和蛋白質不平衡和從食物中吸收多量有毒的肽以及對肝有毒的石膽酸引起。
8.其他原因的肝硬化
(1)先天性酶缺乏:抗ɑ1-胰蛋白酶缺乏症(ɑ1-antitrypsin deficiency, ATɑ1-AT)。ɑ1-AT為糖蛋白,是ɑ1球蛋白的主要組成部分。此病為常染色體顯性遺傳病。正常人血清ɑ1-AT為2.3mg/ml,病人只有(0.2~0.4)mg/ml。ɑ1-AT缺乏引起肝硬化的原因尚未明,推測可能ɑ1-AT對肝細胞有毒性作用,或使肝細胞對毒物的耐受性減低。肝髒病變為大結節或小結節性肝硬化,在肝細胞粗面內質網中(ɑ1-AT的產生部位)有糖蛋白沉積。肝細胞內有PAS染色陽性的包涵體,對診斷有意義。
先天性1-磷酸半乳糖尿甙酸轉移酶(galactose-1-phosphate-uridyl-transferase)缺乏症是引起小兒半乳糖血症的一種少見病。常見嬰兒出生後數月出現肝硬化。肝髒有嚴重的脂肪浸潤及活躍的再生現象,可形成大結節性肝硬化及腹水和門脈高壓症。發病機制尚不清楚,可能與肝內的1-磷酸半乳糖積聚有關。
糖原累積症(glycogen storage disease)可以發生小結節性肝硬化,特別是Ⅲ型多見,與澱粉-1,6-糖甙酶缺乏有關。
(2)代謝障礙性肝硬化:肝豆狀核變性(hepato-lenticular degeneration)又稱wilson病,是一種常染色體隱性遺傳的銅代謝障礙所引起的肝硬化和腦變性疾病。由於大量銅鹽沉積於肝髒引起肝組織的損害,肝髒通常縮小,質地堅硬,屬大結節性肝硬化。
血色病(hemochromatosis):為一罕見的代謝病,系常染色體隱性遺傳病。在基因失常的基礎上有鐵代謝紊亂以致小腸吸收鐵過多,鐵質沉積於肝、胰、心、腎、脾、皮膚等引起細胞破壞、纖維組織增生及髒器功能損害,表現皮膚色素沉著、糖尿病和肝硬化。
(3)遺傳性出血性毛細血管擴張症(hemorrhagic telangiectasia):系常染色體顯性遺傳病。肝硬化為此症的一部分,在肝髒的纖維隔中可見大量的擴張的薄壁毛細血管。
胰腺囊性纖維化(pancreatic fibrocystic disease)為全身性黏液分泌異常,可引起肝髒脂肪浸潤,異常的黏液阻塞胰管,也引起膽管阻塞,形成膽汁性肝硬化。此外尚有先天梅毒也可引起肝硬化。
(二)發病機制
1.病理過程 肝硬化的病因很多,其形成途徑和發病機制亦不相同,有的通過慢性肝炎的途徑(如病毒性肝炎和中毒性肝炎);有的以大囊泡性肝脂肪變性途徑(如酒精性肝病);有的以長期肝內、外膽汁淤積或肝靜脈回流障礙,致門脈區或小葉中央區纖維化的途徑等。不論何種病因、哪種途徑,都涉及到肝細胞炎性壞死,結節性肝細胞再生和肝纖維化等3個相互聯系的病理過程。
(1)肝細胞炎性壞死:肝髒在長期或反復的生物、物理、化學、代謝產物或免疫損傷等病因作用下,均可發生彌漫性肝細胞變性壞死,肝小葉結構破壞、塌陷。若炎症、壞死持續不斷,各種炎性細胞浸潤,將釋放各種細胞因子,促進細胞外間質尤其是膠原蛋白的生成增多。因此,肝細胞炎性壞死不單是肝硬化發生和發展的始動因素,而且貫穿整個病變過程。
(2)肝細胞再生:肝細胞再生是對肝損傷後的修復代償過程。但由於肝小葉纖維支架斷裂或塌陷,再生肝細胞不能沿原支架按單細胞索輪狀排列生長,形成多層細胞相互擠壓的結節狀肝細胞團(再生肝結節)。結節周圍無匯管區,缺乏正常的血循環供應,再生肝細胞形態大小不一,常有脂肪變性或萎縮。再生結節可壓迫、牽拉周圍的血管、膽管,導致血流受阻,引起門靜脈壓力升高。
(3)肝纖維化和假小葉形成:肝纖維化系指肝細胞外的間質細胞(貯脂細胞、成纖維細胞、炎性免疫效應細胞等)增生和細胞外間質成分生成過多、降解減少,致在肝內大量沉積。細胞外間質包括膠原(Ⅰ、Ⅲ、Ⅳ、Ⅴ、Ⅵ型)、糖蛋白(纖維連接蛋白、層粘連蛋白)和蛋白多糖(硫酸軟骨素、硫酸皮膚素、透明質酸)3類大分子組成,分布於肝髒間質、肝細胞及血管的基底膜。Ⅰ、Ⅲ型膠原分布於匯管區,Ⅳ型位於小葉血管、膽管的基底膜、Ⅴ型位於肝血窦周圍和門脈區;纖維連接素、層粘連蛋白與透明質酸等是細胞外非膠原蛋白成分,具有連接和固定作用與膠原相互連接,形成網狀結構,影響了肝髒細胞成分的基因表達。肝髒在肝炎病毒、酒精及其中間代謝產物乙醛、血吸蟲卵、缺氧或免疫損傷等作用下,引起急性、慢性、炎症壞死、激活單核-巨噬細胞系統產生各種細胞因子如血小板源生長因子、轉化生長因子、腫瘤壞死因子、IL-1等,作用於貯脂細胞、成纖維細胞,促其分化增生並分泌、生成大量膠原纖維。各型膠原比例與分布發生變化,Ⅰ/Ⅲ型膠原比值增加。大量Ⅰ、Ⅳ型膠原沉積於Disse腔,使肝窦內皮細胞間“窗”的數量和大小縮減,甚至消失。形成肝窦“毛細血管化”,導致門脈壓力增高,同時妨礙肝細胞與肝窦間營養物質的交換,進一步加重肝細胞的損害。增生的膠原纖維組織自匯管區-匯管區或匯管區-中央靜脈延伸擴展,形成纖維間隔,不僅包繞再生肝結節,並將殘存的肝小葉(一個或幾個)重新分割,改變成為假小葉,形成肝硬化的典型形態變化。假小葉內的肝細胞沒有正常的血循環供應系統,在炎症持續作用下,又可引起肝細胞再壞死及膠原纖維增生。如此反復發展,假小葉形成越來越多,病變不斷加重,導致肝內、外血循環障礙及肝能日益惡化。
2.病理分類 肝硬化因病因、炎症程度以及病情發展的不同,可呈現不同的病理類型,目前仍多采用1974年國際肝膽會議所確定的病理分類,按結節大小、形態分為4型。
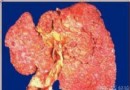
(1)小結節性肝硬化:結節大小比較均勻,一般在3~5mm,最大不超過1cm,纖維隔較細,假小葉大小一致。此型肝硬化最多見。
(2)大結節性硬化:結節較粗大,且大小不均,直徑一般在1~3cm,以大結節為主,最大直徑可達3~5cm,結節由多個小葉構成,纖維隔寬窄不一,一般較寬,假小葉大小不等。此型肝硬化多由大片肝壞死引起。
(3)大小結節混合性肝硬化:為上述二型的混合型,大結節和小結節比例大致相等。此型肝硬化亦甚多見。
(4)不完全分隔性肝硬化:又稱再生結節不明顯性肝硬化,其特點為纖維增生顯著,向小葉內延伸,然肝小葉並不完全被分隔;纖維組織可包繞多個肝小葉,形成較大的多小葉結節,結節內再生不明顯。此型的病因在我國主要為血吸蟲病。
國外有人對520例肝硬化進行病理分類,大結節型達58.8%,以大結節為主的混合型占12.2%,小結節型占9.2%,小結節為主的混合型6.7%,大小結節相等的混合型12.2%,我國仍以小結節性肝硬化多見。同濟醫院51例肝硬化屍檢中,小結節性肝硬化32例,大結節性肝硬化僅2例。梁伯強等報告80例肝硬化屍檢結果,小結節型58.75%,大結節型為23.75%。在一些病例中,上述分類並非固定不變,小結節性肝硬化通過再生改建,可轉變為大結節性或混合性肝硬化。病因與形態學改變有一定相關性,如乙肝性肝硬化常見嗜酸性小體,但也見於酒精性肝硬化;脂肪變性和Mallory小體常見於酒精性肝硬化,也見於Wilson病等;黃色瘤樣變見於膽汁性肝硬化;PAS陽性小體則見於ɑ1-AT缺乏。
3.病理生理 肝硬化時病理生理變化廣泛復雜,幾乎累及全身各個系統髒器。這裡僅就肝硬化時血循環動力學改變加以介紹。
(1)門脈阻性充血與肝內、外分流:在如前所述的各種致病因素的長期作用下,肝實質及其毛細血管網遭到全面破壞與改建。再生肝結節可壓迫其周圍的門靜脈和肝靜脈分支,使血管狹窄、中斷或閉塞;纖維隔的異常增生與瘢痕收縮以及Disse間隙的儲脂細胞轉變為成纖維細胞後,生成大量的膠原纖維,致使肝窦毛細血管化,也是門脈系統阻力增加的重要因素。門脈分支血流進入肝窦時發生淤滯,窦後肝靜脈流出道亦同樣受阻,逐漸形成門靜脈高壓。
由於門靜脈血流阻性充血,在門脈系統引流范圍內的所有髒器均受到影響,如脾髒充血腫大,胃腸充血水腫,胰腺、膽囊亦有相應變化。嚴重者影響這些髒器的功能,並可發生不同程度的形態學變化。隨病情進展,門脈阻塞性充血可改變門脈血流方向,出現逆肝血流,肝髒亦由門脈血液供應為主轉變為以肝動脈供血為主。而肝髒血流量依然減少,可從正常占心輸出量的25%減少至13%。
門脈阻性充血時,肝窦內壓升高,使肝窦內液體成分大量進入窦周間隙,因而形成大量淋巴液。經肝門淋巴結、乳糜池、胸導管引流量太大,可引起淋巴管破裂形成乳糜性腹水;經肝包膜淋巴管吻合支,自肝包膜表面漏入腹腔,可形成腹水;經橫膈淋巴管,流經縱隔或胸膜,影響胸膜淋巴回流,則形成胸腔積液。
門脈高壓經過一定時間達到一定程度時,即會出現肝內、外分流,這種分流為機體的代償機制,以分流門脈系統的阻性充血。肝內分流是纖維隔中的門靜脈與肝靜脈之間的交通支,使門脈血流繞過肝小葉,通過該交通支,進入肝靜脈。肝外分流則位於平時閉合的門-腔靜脈系統間交通支。這些交通支逐漸擴張開放,形成側支循環,部分門靜脈血流經交通支進入腔靜脈,回流入心髒。常見的側支循環有以下幾組:
①門靜脈系統之胃冠狀靜脈與腔靜脈系統之食管靜脈、奇靜脈、肋間靜脈交通支開放擴張,形成胃底與食管靜脈曲張。
②出生後閉合的臍靜脈與臍旁靜脈於門靜脈壓力過高時重新開放,經腹壁靜脈、乳內靜脈進入上腔靜脈,形成臍周與腹壁靜脈曲張。
③門脈系統的直腸上靜脈與腔靜脈的痔中靜脈及痔下靜脈形成痔靜脈擴張。
④腹膜後門靜脈與下腔靜脈之間有許多細小分支相連(Retzius靜脈)。
⑤門靜脈可經脾靜脈、胃靜脈、胰靜脈、左腎上腺靜脈與左腎靜脈溝通。
此外,在肝髒上面無腹膜覆蓋處有許多門靜脈小支與膈靜脈吻合交通。近年來,文獻報道除食管、胃底以外的腸道靜脈曲張,稱異位靜脈曲張,包括十二指腸、空腸、回腸、結腸、直腸,甚至腹腔、盆腔、膀胱、陰道均可發生靜脈曲張形成分流。最具臨床意義的是食管、胃底靜脈曲張,其破裂出血是肝硬化門靜脈高壓症最常見的並發症及致死原因。異位靜脈曲張相對少見,其破裂出血見於十二指腸、結腸,偶見有腹腔內出血者,可造成臨床診斷上的困難。
肝硬化時門脈血流的肝內、肝外分流,使肝細胞對各種物質的攝取、利用、代謝以及庫普弗細胞的攝取、降解、封閉作用明顯減弱,進而使大量有害物質或毒素尤其是肝髒攝取率高、正常情況不進入或極少進入體循環的物質進入全身循環,從而引發一系列病理生理現象,如內毒素血症、高氨血症、高膽酸血症、氨基酸失衡、菌血症及自發性腹膜炎、胰高糖素血症以及腸源性肽類物質血濃度增高等,造成一系列繼發性的病理生理改變及某些藥物(如普萘洛爾)體內半衰期延長。
(2)內髒主動充血與高動力循環:動物實驗研究證明,體液因素在內髒高動力循環發生機制中起重要作用。為此,Benoit提出了舒血管活性物質分流假說。來源於胃、腸、胰腺的血管活性物質很多,由於它們在正常肝髒的攝取率很高,因此肝髒病變以及門靜脈分流時,這些血管活性物質在肝內攝取減少,並大量進入體循環。目前對胰高糖素、一氧化氮、膽汁酸、降鈣素基因相關肽、血管活性腸肽、甲狀旁腺素、前列環素、異亮氨酸、組氨酸肽、P物質等研究較多。Thomas等研究表明,在肝硬化門脈高壓內髒高動力循環中,胰高糖素的作用占30%。研究還表明,膽汁酸具有強烈擴張腸血管作用。同濟醫院近年來對一氧化氮在肝硬化高動力循環中的作用進行了系統研究,結果證實,肝硬化鼠血管產生一氧化氮增多,血漿一氧化氮濃度升高,並與高動力循環有關實驗參數相關,而一氧化氮合成酶抑制劑則可改善高動力循環狀態。該研究還提示,內毒素可能通過誘導一氧化氮合成酶的合成,增加一氧化氮的產生和釋放,而參與肝硬化門脈高壓內髒高動力循環。此外還證實內髒血管床對縮血管活性物質的敏感性降低以及舒血管活性物質對縮血管活性物質的拮抗作用亦參與了內髒充血及高動力循環。據報道,胰高糖素具有拮抗去甲腎上腺素、血管緊張素、血管升壓素等作用。
有人觀察到肝硬化早期腎髒即有鈉水潴留,致血漿容量增加,參與內髒充血及高動力循環。鈉水潴留可能與以下機制有關:
①肝功能減退,抗利尿激素、醛固酮、雌激素等在肝內滅活作用減弱。
②門脈阻性充血時,有效血容量不足,致心房肽分泌減少,同時肝髒合成心房肽亦減少。
③肝髒合成和釋放舒緩素原減少,致擴張血管、調節腎髒血流的緩激肽生成降低。
④腎髒合成前列腺素(舒張血管)不足等與腎髒排鈉障礙有關。
Arroyo研究認為,舒血管物質引起小動脈擴張為腎功能異常的始動因素。由於阻力血管相對充盈不足,腎髒代償性鈉水潴留,以增加血漿容量,當這一代償機制仍不足以維持血循環穩定時,則出現內源性神經激素縮血管物質系統持續激活,以維持血壓,但該系統激活有損於腎髒的灌流量及濾過率,鈉水潴留進一步加劇。內髒主動充血及高動力循環是肝硬化門脈高壓症的結果,也是門脈高壓持續存在的原因之一,並加重肝內外分流。
(3)動靜脈短路與有效血漿容量減少:在舒血管活性物質作用下,不僅擴張內髒小動脈,對外周皮膚、肌肉小動脈亦有擴張作用,使外周血管阻力降低、血容量相對不足。肝硬化時血漿容量增加,但隔離於內髒血管床,因而有效血漿容量減少。此外,毛細血管前括約肌在舒血管活性物質作用下開放,形成動-靜脈短路。這些病理生理變化導致全身各髒器血循環動力學改變。
①心輸出量增加:由於周圍血管阻力降低,有效血容量相對不足,中心靜脈和平均動脈壓降低,為代償此種血流動力學障礙,而出現心輸出量和心髒指數升高,循環時間縮短。臨床表現為心動過速、心髒收縮期雜音、心肌可肥大,但極少發生心功能不全。
②肺內動-靜脈分流與低氧血症:對失代償期肝硬化患者進行血氣分析,常可發現動脈血氧飽和度與動脈氧分壓降低及換氣過度所致的低碳酸血症。這些主要與肝硬化時肺內血循環動力障礙有關。放射學及屍檢證明,肝硬化時肺內常有動-靜脈瘘形成。Martine等對有循環異常的肝硬化患者持續靜脈滴注組胺時發現,肺內以及周圍動-靜脈分流量顯著增加,肺泡-動脈氧差值增大。現多認為,低氧血症主要與肺內及(或)周圍血管的動-靜脈分流有關;其他原因尚有氧離解曲線右移、肺彌散-灌注比例失調及肺通氣灌注比率異常。
肝硬化時肺循環異常尚包括肺動脈高壓,其原因可能與門脈與肺內動脈之間存在分流,使腸源性毒素如內毒素、組胺等進入肺動脈,引起肺動脈收縮以及壓力較高的門脈血流直接注入肺動脈等有關。
③腎髒血流動力學改變:腎功能損害與肝硬化的病變程度相關。代償期肝硬化時腎血漿流量(RPF)與腎小球濾過率(GFR)正常。伴有腹水,尤其是頑固性腹水或並發肝腎綜合征時,其RPF和GFR均有中至重度降低。雖然腎功能有嚴重障礙,但病理形態學卻無特殊改變。
腎血流量減少是產生RPF和GFR異常的病理生理基礎。腎血流量減少的機制可歸納為:有效循環血容量不足;腎血管收縮;腎血流由皮質向髓質部分流等。